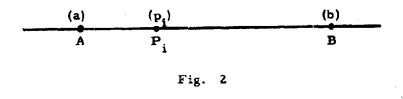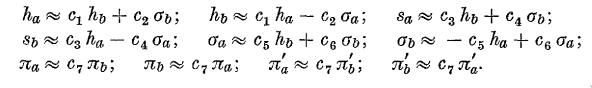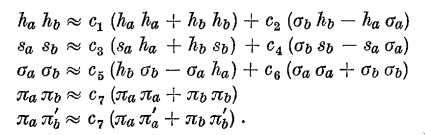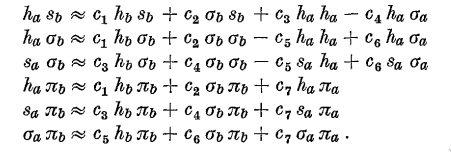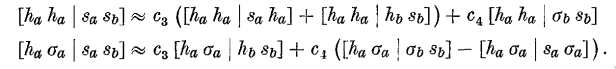While DF is still an important avenue for speeding calculations
today, a couple of factors made it invaluable at the advent of
computational chemistry. The first of these was the use of Slater
type orbitals (STOs) in early chemistry software. STOs are close
approximate solutions of the Schrödinger equation and as such were a
clear choice for early basis sets. Despite this accuracy, the great
shortcoming of STOs is that products of more than two STOs lead to
expressions that are impossible to integrate analytically. This is
in contrast to the most popular type of orbital used now, the
Gaussian type orbital (GTO), which when multiplied with another GTO
gives another translated GTO, which in turn can be readily
integrated analytically. This necessitated the use of approximation
techniques or semi-empirical parameters in STO calculations
involving more than two centers. The other big factor is a bit more
obvious but still important: old computers were slower and more
expensive to operate. This means that numerical integrals were often
too expensive or time-consuming to compute, which compounds the lack
of analytic forms for the multi-center STO integrals.
With this in mind, one of the earliest studies on a triatomic
molecule necessitated the development of an early DF technique that
involved expanding two-center charge distributions by a
least-squares procedure as a sum of single-center charge
distributions centered on a line passing through the two
centers. This sounds complicated, but the main idea is summarized
readily by Fig. 2 from the paper Boys and Shavitt (1959) reproduced
below:
Similarly, the corresponding mathematical expression is given by the
equation
\begin{equation}
ab \cong \sum_{i}^m C_i p_i,
\end{equation}
where the \(p_i\) are a uniform set of 20 symmetrically placed
exponential functions and the \(C_i\) are coefficients adjusted to
minimize the metric
\begin{equation}
\int(ab - \sum_p C_{i}p_i)d\tau,
\end{equation}
which is just the previous equation rearranged and integrated over
the set of two-center charge distributions \(ab\).
Basically, the authors approximate the difficult integrals by a
linear combination of primitive exponential functions. If you are
familiar with the STO-nG basis sets, and really any modern Gaussian
basis sets, this should sound quite familiar. The authors are
essentially describing a STO-20E basis set, where they approximate
an STO as a contraction of 20 exponential functions rather than n
Gaussian functions. This should serve as a clear indication of the
difficulty of the integrals themselves since the authors are willing
to increase the number of terms by a factor of 20 to obtain easier
computations.
Seven years later, in 1966, the same problems with STOs persisted,
and many more methods had been proposed to treat the necessary
integrals approximately by people you have probably heard
of. Mulliken, for example, proposed approximating a two-center
orbital product by the simple average of the single-center orbitals
involved. Löwdin made the rather straightforward suggestion to
extend this by taking the weighted average to preserve the correct
dipole moment. These and many more are described by Harris and Rein,
but the takeaway is that all of these methodologies can be seen as
taking the leading terms from infinite series expansions of the
two-center charge densities, much like Boys and Shavitt did. The
goal of the authors then was to use this perspective, combined with
a new procedure for optimizing the coefficients of the charge
densities, to yield the most accurate approximation scheme to date.
Their general scheme was to expand orbitals about one center in a
series about the other center using terms of each atomic symmetry,
where at least in this paper they have restricted themselves to
\(s\) and \(p\) symmetry as you can see in the figure below. They
use \(h\) to denote a 1\(s\) orbital, \(s\) for 2\(s\), \(\sigma\)
for the \(m=0\) 2\(p\) orbitals, and \(\pi\) for the \(m=\pm1\)
2\(p\) orbitals. The prime indicates the negative version.
Using these definitions, the products of the orbitals can be written
as
and
The rather primitive fitting is done by taking pairs of integrals
involving the same coefficients and solving the resulting linear
system, as shown in the example below, where \(c_3\) and \(c_4\) are
being determined.
Once the coefficients are determined, the difficult, four-center
electron repulsion integrals can then be computed as linear
combinations of the much easier one-center integrals and two-center
Coulomb integrals. As the authors point out, this procedure requires
integrals on only \(\frac{n(n-1)}{2}\) pairs of centers compared to
the \(n^4\) needed for the full computation, and they achieve
accuracies relative to the exact integral calculations of 1 kcal.
By 1971 the use of GTOs had become more widespread following the
introduction of the STO-nG basis sets by John Pople in 1969, but
there are some downsides of using GTOs as well. The chief among
these is the fact that GTOs have improper limiting behavior at both
the nucleus, where they are rounded instead of forming a cusp, and
at long range, where they decay too quickly. However, these issues
can be mitigated by taking linear combinations of multiple primitive
Gaussian functions to form something more closely resembling an
STO. As such, most modern software instead uses this type of GTOs
where multiple individual Gaussians are contracted into something
that can more closely emulate the limiting behavior of STOs. This
practice is most obvious in the naming of the aforementioned STO-nG
basis sets, which form approximate STOs from contractions of n
primitive (G)aussian functions. Another problem is that this
contraction scheme introduces a potentially large number of GTOs
relative to the number of STOs required, but now at least the
integrals can be evaluated analytically. Nevertheless, numbers were
larger in the 1970s relative to the available computer power, so
reducing the number of integrals by decomposing them into reusable
pieces was the problem Billingsley and Bloor sought to address in
their work on the limited expansion of diatomic overlap (LEDO)
approximation. While this may sound somewhat different from the
earlier problem, this begins to illustrate the wide application of
the density fitting technique.
Another deficiency Billingsley and Bloor pointed out in the earlier
work, including that of Harris and Rein, was the lack of full
symmetry handling in the approximate two-center charge
distributions. For example, in the Harris and Rein formulation,
using only one 1\(s\) and 2\(p\) orbital for the approximations
leads to missing irreducible representations of the rotation groups
for the orbitals with \(p\) symmetry. This leads to incorrect or
missing nodal structures in the resulting approximate
distributions. To remedy this, Billingsley and Bloor suggested the
following definition for the two-center charge distribution
\(\pi_k^{AB}\):
\begin{equation}
\pi_k^{AB} \cong \sum_p^{\text{on }A}C_{kp}\Omega_p^A
+ \sum_q^{\text{on }B}C_{kq}\Omega_q^B
\end{equation}
where \(\Omega_p^A\) is a unique one-center distribution formed by
the product of two orbitals, \(\chi_m^A\chi_n^A\). This gives a set
of approximate two-center charge distributions as a linear
combination of the one-center distributions inherent in the basis
set of choice since we are summing over the unique sets of
one-center distributions on the two atomic centers in question. This
has the additional benefit of generalizing immediately to any set of
AO basis functions. With this formulation in hand, the authors now
needed to determine the \(C\) values. Because of the success of the
Harris-Rein approach there, they maintained the same approach of
fitting the coefficients to a relatively small set of exact
integrals but introduced a matrix notation more appropriate for the
larger systems of equations involved.
The fundamental equation here is
\begin{equation}
(\Omega_i^A | \frac{1}{r_{12}} | \pi_k^{AB}) \cong
\sum C_{kp}(\Omega_i^A| \frac{1}{r_{12}} | \Omega_p^B),
\end{equation}
where you can use the definitions
\begin{equation}
L_{ij} \equiv (\Omega_i^A | \frac{1}{r_{12}} | \pi_j^{AB})
\end{equation}
and
\begin{equation}
J_{ip} \equiv (\Omega_i^A| \frac{1}{r_{12}} | \Omega_p^B),
\end{equation}
to rewrite the problem as the matrix expression
\begin{equation}
\boldsymbol{L} = \boldsymbol{CJ},
\end{equation}
which can be solved for the coefficients by inverting
\(\boldsymbol{J}\). The authors point out that this inversion can be
quite touchy due to the often ill-conditioned \(\boldsymbol{J}\)
matrix, so care must be taken in performing it. However, the
resulting coefficients can be used in Eqn. 4 to yield the full set
of two-center charge distributions, which in turn are used to
generate three- and four-center integrals by taking linear
combinations of the one- and two-center Coulomb and hybrid
integrals. The agreement produced by the LEDO method is only 12.5
kcal/mol relative to full integral calculations, but as the authors
point out, this cannot be compared directly to the results of Harris
and Rein, who only looked at homonuclear diatomics, whereas
Billingsley and Bloor examined a test set including polyatomic
molecules like formaldehyde, methane, acetylene, and ethylene.